It is a non-curable condition with an average life expectancy of 35 to 40 years, but treatments can alleviate the symptoms. Discover what Cystic Fibrosis is.
Cystic Fibrosis is a genetic disease that affects approximately 75,000 people worldwide, with a higher incidence in the European Union. In Portugal, it is estimated that 30 to 40 children are born with this disease each year. Despite being rare, it is the most common genetic disease in the Caucasian population. Find out the signs to watch for and how to improve your quality of life.
What is Cystic Fibrosis?
Cystic Fibrosis is a severe and rare genetic disease that hinders the normal production of a protein called CFTR (Cystic Fibrosis Transmembrane Regulator). Since this protein is responsible for transporting water and minerals between cells, this disease causes excessively salty sweat and thick, sticky mucus.
In milder mutations, this protein is still synthesized but does not function properly. In more severe mutations, the protein is not even produced, leading to an earlier onset of the disease. As a genetic disease, Cystic Fibrosis is inherited from parents and typically manifests in childhood. However, for a child to have Cystic Fibrosis, both the mother and father must be carriers of the genetic mutation. Additionally, factors such as tobacco, pollution, or allergens can accelerate the progression of Cystic Fibrosis.
Cystic Fibrosis: symptoms
The consequences of Cystic Fibrosis affect the entire body, with symptoms that can occur in any organ of the human body. However, the most affected areas are the digestive and respiratory systems, which can later compromise the pancreas, intestine, or liver. The main manifestations of the disease include:
- Respiratory infections, bronchitis, and shortness of breath due to the presence of thick and difficult-to-clear secretions.
- Intestinal obstructions, greasy stools, or malnutrition due to the pancreas's inability to produce digestive enzymes.
- Production of sweat with a high salt content, associated with the risk of dehydration due to low sodium levels.
- Male infertility caused by the obstruction of the sperm-carrying channels.
Cystic Fibrosis typically appears in early childhood and can vary in severity depending on the extent to which organs are affected.
How is Cystic Fibrosis diagnosed?
The diagnosis of Cystic Fibrosis is made when there is a clinical suspicion of the disease in a medical consultation setting. Two types of tests are used:
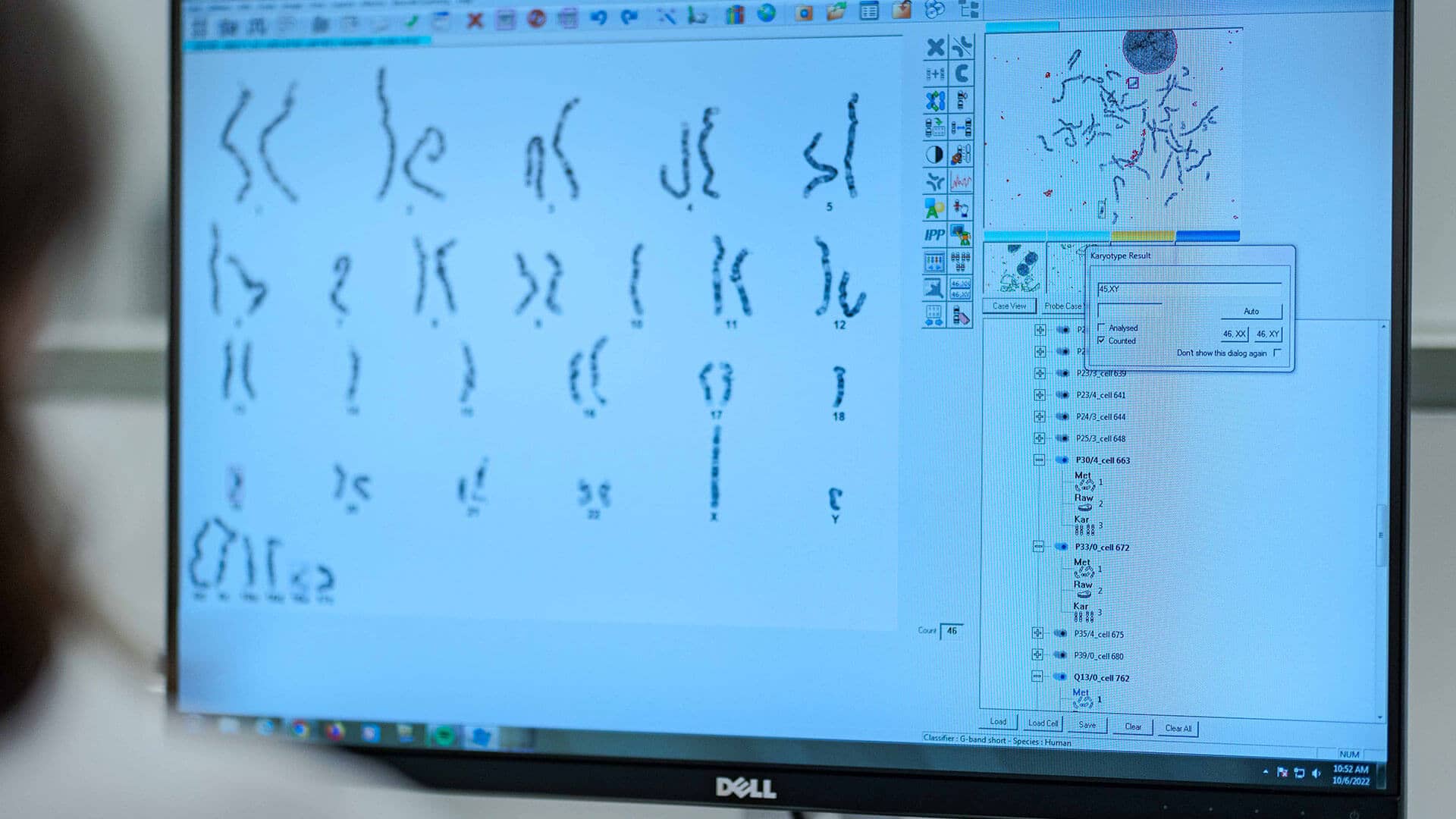
- Sweat test: In the initial screening, a test measuring chloride levels in sweat can be performed. Elevated levels indicate a positive diagnosis. After two positive sweat tests, a genetic test is conducted.
- Genetic test: A blood sample is taken to search for mutations in the CFTR gene. Initially, the most common mutations are investigated, and, if necessary, complete sequencing is performed.
It is also possible to conduct prenatal screening from the 12th week of gestation, when one member of the couple is at risk of being a carrier of the mutation. Furthermore, since 2013, neonatal screening for Cystic Fibrosis has been included in the National Early Diagnosis Program, known as the "heel prick" test.
What is the treatment for Cystic Fibrosis?
Cystic Fibrosis does not yet have a cure, but significant reduction of disease-related complications is possible when detected early. Timely clinical intervention allows for considerable improvements in the quality of life and increased life expectancy for patients (currently ranging between 35-40 years of age).
The therapies used primarily aim to control symptoms, taking into account considering the patient's age and the progression of the condition. Treatment is targeted at the affected organs and may include antibiotic, anti-inflammatory, bronchodilator, or mucolytic medications. In some cases, nutritional supplements may be recommended.
As a complement to drug therapy, respiratory rehabilitation may be necessary to facilitate secretion drainage. In severe cases, surgical procedures, including lung transplantation, may be considered. The Specialist Doctor may also recommend certain lifestyle habits to help slow the progression of Cystic Fibrosis, such as adjusting salt intake, increasing hydration, regular hand hygiene, or engaging in physical exercise as regularly as possible. All recommendations should always be prescribed by the Specialist Doctor.
Genetic Counseling at Joaquim Chaves Saúde
The rarity, complexity, and specificity of Cystic Fibrosis treatment require specialized multidisciplinary teams to provide appropriate care for children, young people, or adults. The objective is to assess personal and family risk and provide the necessary clinical guidance to individuals who are already carriers or at risk of developing the disease.
At the Medical Genetics Consultation at Joaquim Chaves Saúde, it is possible to obtain a clinical diagnosis of Cystic Fibrosis and receive appropriate genetic counseling. Here, you will find excellent healthcare, ensuring access to the most advanced methods of Cystic Fibrosis treatment. Don't delay any longer and schedule your appointment now.



